Quest for the right Drug
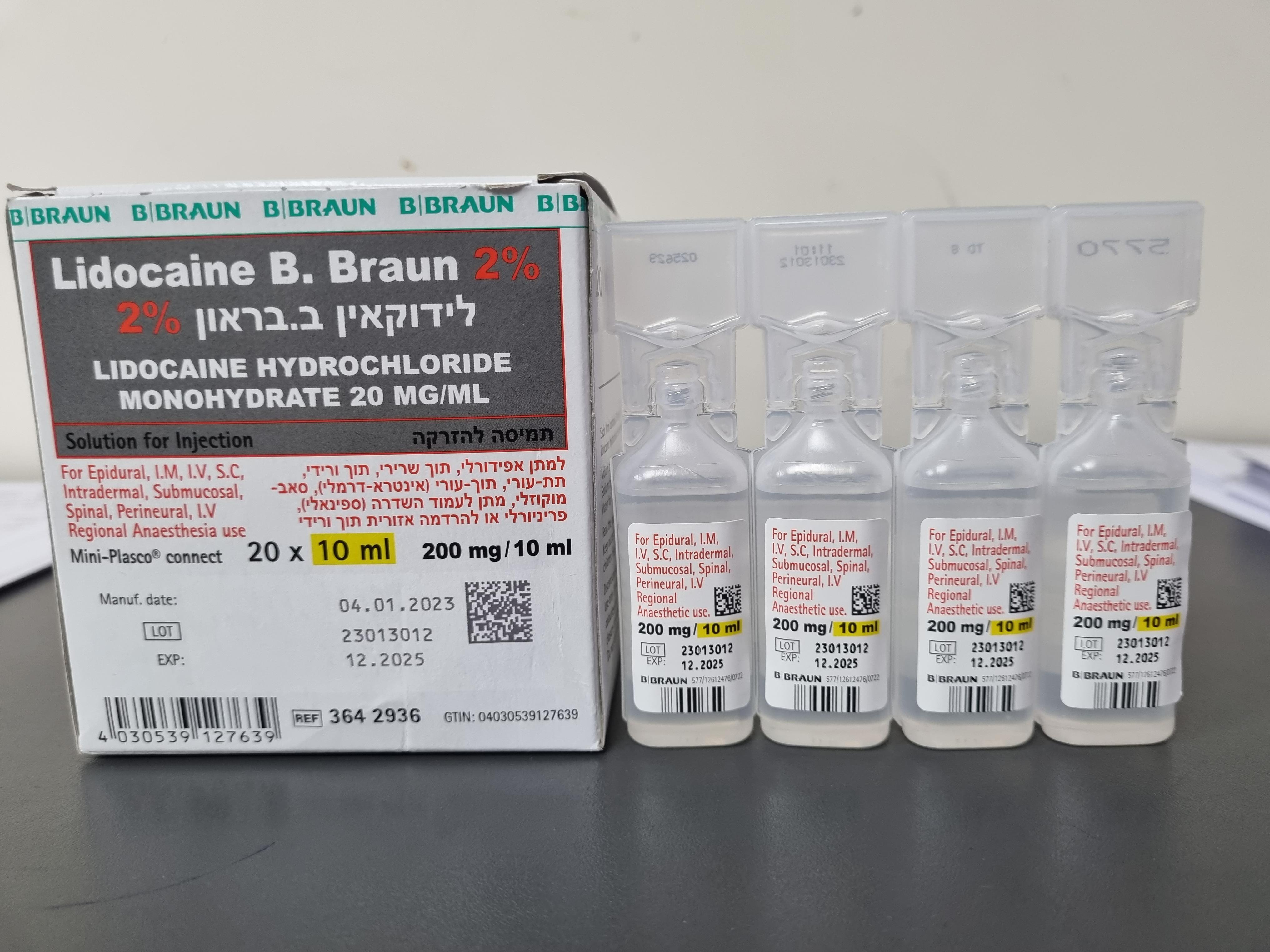
לידוקאין ב.בראון 2% LIDOCAINE B.BRAUN 2% (LIDOCAINE HYDROCHLORIDE MONOHYDRATE)
תרופה במרשם
תרופה בסל
נרקוטיקה
ציטוטוקסיקה
צורת מתן:
תוך-ורידי, תוך-שרירי, אפידורל, תת-עורי, תוך-עורי, הזרקה תת-רירית, שדרתי, לסביבת העצב, : I.V, I.M, EPIDURAL, S.C, INTRADERMAL, SUBMUCOSAL, SPINAL, PERINEURAL, I.V REGIONAL ANAESTHESIA
צורת מינון:
תמיסה להזרקה : SOLUTION FOR INJECTION
עלון לרופא
מינוניםPosology התוויות
Indications תופעות לוואי
Adverse reactions התוויות נגד
Contraindications אינטראקציות
Interactions מינון יתר
Overdose הריון/הנקה
Pregnancy & Lactation אוכלוסיות מיוחדות
Special populations תכונות פרמקולוגיות
Pharmacological properties מידע רוקחי
Pharmaceutical particulars אזהרת שימוש
Special Warning עלון לרופא
Physicians Leaflet
Pharmacological properties : תכונות פרמקולוגיות
Pharmacodynamic Properties
5.1 Pharmacodynamic properties Pharmacotherapeutic group Anaesthetics, local, amides: ATC code: N01B B02 Antiarrhythmics, class Ib: ATC code: C01BB01 Mechanism of action Local and regional anaesthesia Lidocaine is a local anaesthetic agent of the amide type. Lidocaine reduces the permeability of cell membranes for cations, in particular sodium ions, at higher concentrations also for potassium ions. This leads, depending on the concentration of lidocaine, to reduced excitability of the nerve fibres because the increase of sodium permeability producing the action potential is slowed down. From inside the cell the lidocaine molecule enters the open sodium channel and blocks it by binding to a specific receptor. A direct effect of incorporation of lidocaine in the cell membrane is much less relevant. Because lidocaine, before reaching its site of action, must pass into the cell, its effect depends on its pK a and on the environmental pH, i.e. on the proportion of the free base which is the moiety predominantly migrating through the lipophilic membranes of nerve fibres. In inflamed tissue the local anaesthetic effect is reduced due to the lower pH in such regions. Antiarrhythmic therapy In membranes of myocardial fibres lidocaine inhibits the large transient increase in the permeability of the membrane for sodium channels during the plateau of the action potential and increases the potassium efflux during repolarisation period. In Purkinye fibres the duration of action potentials and their effective refractory time are shortened while impulse conduction is slowed down. Impulse conduction in the sinus node and supraventricular regions remains virtually unaffected. Clinical efficacy and safety Local and regional anaesthesia Lidocaine inhibits the function of excitable structures such as sensor, motor and autonomic nerve fibres and the cardiac impulse conducting system. Lidocaine reversibly inhibits the conduction in sensitive nerve fibres in the area of application. The order of loss of nerve function is as follows: pain, temperature, touch, and pressure. The local anaesthetic effect of lidocaine lasts for about 30 minutes- 3 hours depending on the type of anasesthesia. Antiarrhythmic therapy In the myocardium the excitation and fibrillation thresholds are raised. Lidocaine suppresses heterotopic pacemakers and action potentials originating from delayed potentials and tachyarrhythmias caused by circus rhythm. The sodium channels more avidly bind lidocaine when the membrane is depolarized. Therefore the antiarrhythmic effect of lidocaine is particularly marked in cases of increased excitation frequency The effect of lidocaine is enhanced if the resting potential is less negative, e.g. in hyperkalaemia and/or myocardial ischaemia. In situations of hyperpolarisation, e.g. due to hypokalaemia, the effect of lidocaine is reduced. Lidocaine has been shown to eliminate re-entrant ventricular arrhythmias in the late myocardial phase by further depression and blocking of conduction in the re-entrant pathway. Therapeutic plasma concentrations should lie between 1.5 and 5 mg/l. Beyond 5 mg/l, toxic effects on the CNS and the cardiovascular system are to be expected. Other pharmacological effects Lidocaine shows weak parasympatholytic activity. Intradermally administered lidocaine acts at low concentrations as a mild vasoconstrictor and at higher concentrations as vasodilator. Antiarrhythmic therapy The effects of lidocaine on myocardial contractility, blood pressure, cardiac output and heart rate are very small. Patients with impaired function of the sinus node, however, may respond particularly markedly to the conduction suppressing effect of lidocaine. In the period immediately following myocardial infarction the coronary blood flow may be increased by lidocaine. Paediatric population There are no data indicating that the pharmacodynamic properties of lidocaine in children should be different from those established for adults.
Pharmacokinetic Properties
5.2 Pharmacokinetic properties Absorption Plasma levels depend on the site and mode of administration. However, there is a poor relationship between the amount of local anaesthetic injected and peak plasma levels. After intravenous administration the bio-availability is 100 %. Maximum concentrations are achieved within latest 30 minutes, in the majority of patients maximum concentrations are met within 10-20 minutes. After intramuscular injection of 400 mg of lidocaine Hydrochloride monohydrate for intercostal block, the maximum plasma concentration (C max) has been determined to be 6.48 mg/l, attained after 5 – 15 min (t max). After intravenous administration, onset of the therapeutic effect of lidocaine is rapid. Therapeutic plasma concentrations are reached within 1 ‑ 2 min. The effect of a bolus injection lasts for 10 ‑ 20 min; in order to maintain the therapeutic effect of lidocaine, its administration must be continued in the form of an intravenous infusion. After continuous infusion and when no loading dose is given the steady state of plasma concentration was achieved not earlier than 5 hours (range, 5 – 10 hours) of beginning of the infusion. However, therapeutic concentrations had already been achieved after 30 – 60 min. After subcutaneous administration, C max values reached 4.91 mg/l (vaginal injection) or 1.95 mg/l (abdominal injection), respectively. In a study involving 5 healthy volunteers, after maxillar-buccal infiltration anaesthesia with 36 mg of lidocaine, using a 2 % solution, the C max value reached 0.31 mg/l. After epidural injection the measured maximum plasma concentrations do not seem to be directly proportional to the dose applied. Administration of 400 mg resulted in Cmax values of 3 - 4 mg/l. No data are available on pharmacokinetics after intrathecal administration. Distribution Lidocaine follows a biphasic elimination kinetic. After intravenous administration the drug substance is first rapidly distributed from the central compartment into intensively perfused tissues and organs (a-distribution phase). This phase is followed by redistribution into skeletal muscles and adipose tissue. The half life time during the a-distribution phase is approximately 4-8 minutes. Distribution into peripheral tissues is predicted to occur within 15 min. The plasma protein binding rate is approximately 60 – 80 per cent in adults. It is dependant on the drug concentration and additionally on the concentration of the α-1-acid glycoprotein (AAG). The AAG is an acute phase protein that is binding free lidocaine and may be increased e.g. after trauma, surgery or burns depending on the pathophysiological condition of the patient. To the contrary it had been shown that AAG concentrations are low in neonates and patients suffering from liver impairment leading to a marked reduction of lidocaine plasma protein binding. The distribution volume may be altered in patients suffering from further diseases, e.g. heart insufficiency, liver insufficiency or renal insufficiency. Biotransformation Besides distribution of Lidocaine in other compartments (e.g. cerebrospinal fluid), the drug rapidly metabolised in the liver by mono- oxygenases mainly via oxidative desalkylation, hydroxylation at the aromatic ring and hydrolysis of the amide bond. Hydroxylated derivatives undergo conjugation. In total, approx. 90 % of lidocaine is metabolised to 4-hydroxy-2,6-xylidine, to 4- hydroxy-2,6-xylidine glucuronide and to a lower degree to the active metabolites monoethyl glycine xylidide (MEGX) and glycine xylidide (GX). The latter may accumulate during longer lasting infusions or in the presence of severe renal insufficiency due to their longer half life time as compared to lidocaine itself. In the presence of liver diseases the metabolic rate may be reduced to 10 – 50 per cent of normal. Results with human liver microsomes and recombinant human CYP isoforms demonstrated that CYP1A2 and CYP3A4 enzymes are the major CYP isoforms involved in lidocaine N-deethylation. The hepatic blood flow appears to limit the rate of lidocaine metabolism. As a consequence the plasma t1/2 of lidocaine and its metabolites may be prolonged and significant effects on pharmacokinetics and dosage requirements of lidocaine are to be expected in patients with impaired liver perfusion, e.g. after acute myocardial infarction, in the presence of cardiac insufficiency, liver disease or congestive heart failure. Elimination Less than -10 per cent of lidocaine are excreted unchanged in urine, the remaining proportion in the form of the metabolites. The elimination half-life time is 1.5 – 2 hours in healthy adults and approx. 3 hours in newborns. The half-life times of the active metabolites monoethyl glycine xylidide (MEGX) and glycine xylidide (GX) are 2-6 hours and 10 hours, respectively. Since their plasma t1/2 are longer than that of lidocaine, accumulation of metabolites, particularly GX, may occur during prolonged infusion. Additionally, the elimination rate depends on the pH; it can be increased by acidification of the urine. The plasma clearance is about 0.95 ml/min. Paediatric population After epidural anaesthesia of the mother, the elimination half-life time in the new-born was approximately 3 hours; after infiltration of the perineum and after paracervical block lidocaine was found in the urine of the new-born during 48 hours following anaesthesia. The plasma t1/2 is increased 2-3 fold in neonates, due to a slower rate of metabolism and in parts to the expanded distribution volume. Absorption and elimination may be faster in children than adults, although other studies suggested that differences in pharmacokinetics (between children and adults) decrease by correcting for BW. Pharmacokinetics in special clinical situations Renal impairment • In the presence of renal insufficiency the plasma half-life time of lidocaine seemed to be unaltered except for some accumulation of GX during infusion of 12 hours or more. This accumulation seemed to be associated with long-term administration of the drug. However in patients with severe renal insufficiency clearance of lidocaine was approximately halved and half-life time of lidocaine was about twice the amount than in healthy patients. Elderly Elimination half-life and volume of distribution may appear to be prolonged resp. increased in the elderly due to reduced cardiac output and/or hepatic blood flow. Pregnancy and lactation Lidocaine passes across the placental barrier by simple diffusion and reaches the fetus within a few minutes of administration. After epidural administration, the fetal to maternal plasma concentration ratio is 0.5 – 0.7. After infiltration of the perineum and after paracervical block, markedly higher concentrations of lidocaine have been found in umbilical blood.The fetus is able to metabolise lidocaine. The levels in fetal blood are approximately 60% of the concentrations in the maternal blood. Due to a lower plasma protein binding in foetal blood, the concentration of the pharmacologically active free lidocaine is 1.4 fold the maternal concentration Lidocaine is secreted into breast milk only in small amounts.

שימוש לפי פנקס קופ''ח כללית 1994
לא צוין
תאריך הכללה מקורי בסל
לא צוין
הגבלות
לא צוין
מידע נוסף
